BioSolvetIT SeeSAR is a highly interactive, intuitive visualization and analysis software for structure-based drug design (SBDD). Its core philosophy is to provide medicinal chemists with immediate, real-time feedback on how structural changes to a potential drug molecule (ligand) will affect its binding affinity and interactions with a target protein. Users can visually “grow,” “evolve,” and modify molecules directly in the 3D binding site, with SeeSAR instantly calculating and displaying updated binding affinity estimates (HYDE scoring), synthetic accessibility, and potential toxicity flags. This dramatically accelerates the lead optimization cycle.
It acts as a powerful, user-friendly interface that bridges the gap between high-end computational chemistry and the practical, design-focused workbench of a medicinal chemist.
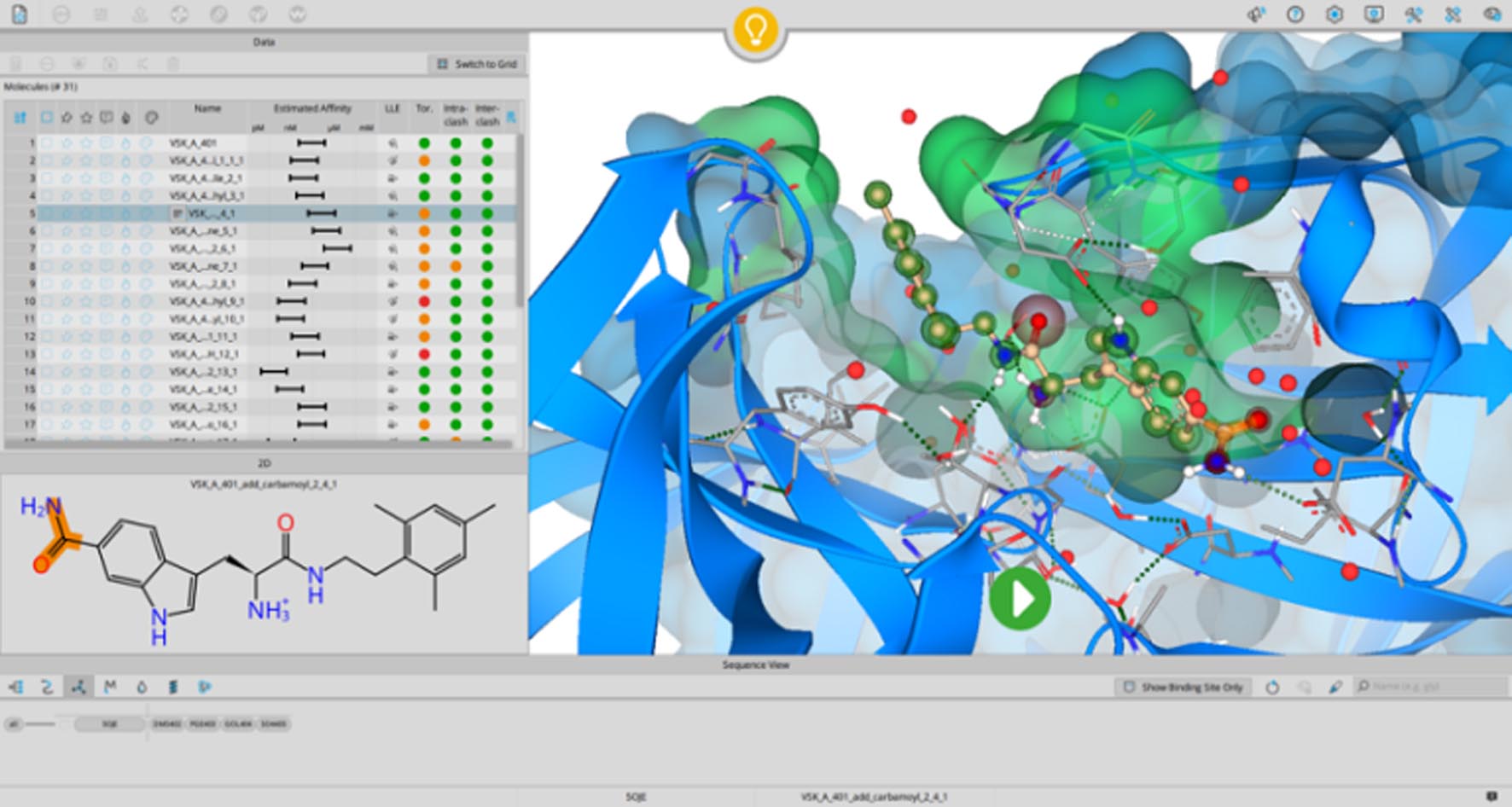
SeeSAR focuses on visual interactivity and real-time computational feedback for drug design.
Real-Time Affinity Estimation (HYDE Scoring):
Instant ΔG Calculation: As you draw or modify a ligand, SeeSAR instantly displays the predicted change in binding free energy using its proprietary HYDE (HYdrogen bond and DEhydration) scoring function, which is based on physicochemical principles.
Visual Energy Contributions: Atoms are color-coded (red = unfavorable, green = favorable) to show their individual contribution to binding, making it clear which parts of a molecule are beneficial or problematic.
Interactive Molecule Editing & Growing:
Sketch-in-3D: Click in the 3D protein active site to add or modify atoms and bonds directly. The software handles geometry optimization in real-time.
Fragment Growing: Start from a core scaffold or fragment and visually “grow” the molecule by adding functional groups from a built-in library, with instant affinity feedback.
Bioisosteric Replacement: Suggests and ranks potential bioisosteric replacements for selected molecular parts to improve properties while maintaining binding.
Integrated Data & Property Analysis:
Property Dashboard: Displays key drug-like properties (cLogP, molecular weight, TPSA, solubility, synthetic accessibility, etc.) that update in real-time with each modification.
Toxicity & Interaction Alerts: Flags potential toxicity risks (e.g., PAINS, unwanted interactions) early in the design process.
Visualization & Analysis:
Advanced 3D Visualization: High-quality rendering of protein-ligand complexes, surfaces (pockets, solvent-accessible surface), and interactions (H-bonds, hydrophobic contacts, π-stacking).
Comparison & R-group Analysis: Easily compare multiple ligands aligned in the binding site to analyze structure-activity relationships (SAR).
Performance & Usability: General speed improvements and interface refinements.
Enhanced HYDE Scoring: Refinements to the affinity estimation algorithm for greater accuracy.
Expanded Fragment & Template Libraries: More building blocks and design ideas readily available.
Improved Data Handling: Better integration with external data sources and file formats.
Advanced Customization: More options for expert users to tailor the analysis and display.
SeeSAR is designed to run efficiently on standard modern workstations, leveraging GPU acceleration for visualization.
Minimum System Requirements:
Windows 10 (64-bit), macOS 10.14+, or a modern Linux distribution (Ubuntu 20.04+, RHEL 8+); a multi-core Intel/AMD processor; 8 GB RAM; a graphics card with OpenGL 4.0 support and 2 GB VRAM; 500 MB free disk space; and a 1920×1080 display.
Recommended for Professional Use:
Windows 10/11, macOS 12+, or Linux; a fast multi-core processor (Intel i7/i9 or AMD Ryzen 7/9); 16-32 GB RAM; a dedicated NVIDIA GeForce/Quadro or AMD Radeon graphics card with 4-8 GB VRAM; an SSD; and a high-resolution display for optimal workspace clarity.
Price: 125 $
Price Currency: $
Operating System: Windows
Application Category: Computational Chemistry













Reviews
There are no reviews yet.